


Recently LGC Clinical Diagnostics had the pleasure to host a webinar, bringing together a panel of academic, clinical, and industry experts in the field of methylated DNA testing, who discussed the promises and challenges of early cancer screening and recent technological advances.
It was a great opportunity to hear from the pioneer of prenatal cfDNA testing, Dennis Lo, MD, PhD, and an expert in computational cancer genomics analysis, Jimmy Lin, MD, PhD, MHS, as well as Yves Konigshofer, PhD, the head of technology development at LGC Clinical Diagnostics.
First, Dr. Lo, Director of the Li Ka Shing Institute of Health Sciences at The Chinese University of Hong Kong, presented his latest research on direct methylation analysis of long cfDNA in plasma. Although cfDNA is generally accepted to consist of mainly short molecules around 170 bp long, over the last 2 years Dr. Lo and his group have been asking whether the reported cfDNA size might be biased by the dominant short read sequencing approach. To answer that they sequenced plasma samples from pregnant women with PacBio long read, single-molecule real-time sequencing and indeed they observed a sizeable population of long molecules, up to the kbp size range. The proportion of these molecules increased during pregnancy, which was significant not only because it indicated they had a biological role, but also because it meant sequencing one such long molecule might be enough to obtain a full fetal or maternal haplotype and information about the tissue of origin (1).
To deduce the methylation profile from long read PacBio data, Dr. Lo’s group developed a holistic kinetic (HK) model based on the pulse delay between the nucleotide signals on both DNA strands. It is based on an AI trained on known methylated and unmethylated molecules and can be directly applied to native DNA without requiring any chemical or enzymatic conversions of DNA prior to sequencing (2). Comparing this method with bisulphite sequencing showed a good level of concordance for different tissues and cell lines.
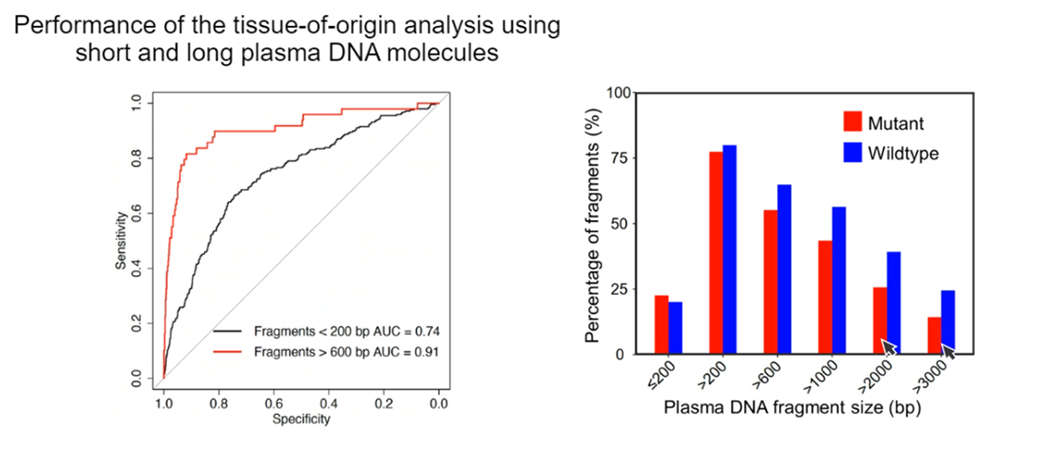
They successfully applied the HK model to interrogate the methylation status of cfDNA and assess the maternal or fetal origin of individual molecules, taking advantage of the abundance of CpG sites on longer molecules which allows for a more reliable estimation of methylation patterns.
The same pattern of a significant population of long cfDNA molecules detected by single-molecule long read sequencing could be seen in plasma of cancer patients, both for wild type and mutation carrying molecules, opening numerous application possibilities for both NIPT and oncology.
Dr. Lo likened the amount of information available from long molecules to being able to read whole text documents sent by email rather than being limited to text message snippets.
Our next speaker was Dr. Jimmy Lin, the Chief Scientific Officer at Freenome. Dr. Lin is leading the development of solutions to early cancer detection and screening based on a multi-omics approach, which is really complementary to the methods highlighted by the first speaker. Their first blood test is focused on colorectal cancer (CRC), CRC is the second deadliest cancer in the US, but highly treatable when caught early, as opposed to, for example, ovarian cancer, for which a recent large clinical trial in the UK showed that early screening offered no improvement in survival (3).
Dr. Lin pointed out that cancer heterogeneity requires combining multiple analytes for effective early detection. Since cancer is not a static disease, the biological signals change during cancer evolution. As a result, different analytes can be more informative at different disease stages. Non-tumor derived signals, especially proteins, have their greatest impact at the earliest phases when there is not enough ctDNA present to be detected and analysed. ctDNA status including methylation becomes more informative in the later stages of cancer.
Freenome’s platform combines proteomics, transcriptomics, genomics and epigenomics. Integration of the information from all these data sets first requires featurization (which is to say what biological information each analyte can provide), followed by a machine learning classifier. In his presentation Dr. Lin focused on methylation, asking what can be done with the methylation state information. First, according to Dr. Lin, there are many different ways of measuring methylation including simple frequency, methylation entropy or methylation haplotyping. How you combine this information and take non-methylation data from genomics into account can provide better cancer detection.
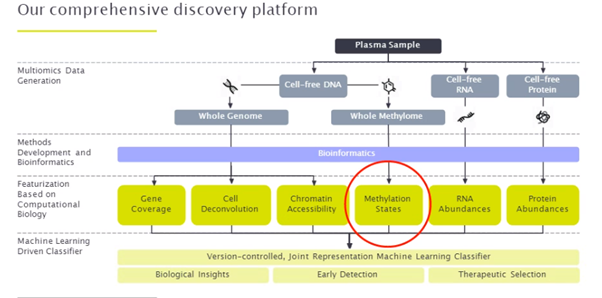
Many features of ctDNA are important for early detection (nucleosome footprint, topology, fragmentomics), but a lot of the data in their test is based on methylation.
In the AI-EMERGE trial, using this approach showed high sensitivity and specificity in early detection of CRC across all stages. Similarly, combining non-tumor and tumor-derived Information allowed for earlier detection of adenomas, and leveraging methylation data together with a well-known CA19-9 protein biomarker enabled achieving a very high specificity in detecting pancreatic cancer in a proof of concept study.
Watch the webinar now by clicking here, or on the button below.

To advance to the next blog article in this series, click here.
References:
- Yu, S. C. Y., et al. (2021). Single-molecule sequencing reveals a large population of long cell-free DNA molecules in maternal plasma. Proceedings of the National Academy of Sciences of the United States of America, 118(50), e2114937118.
- Tse, O. Y. O., et al. (2021). Genome-wide detection of cytosine methylation by single molecule real-time sequencing. Proceedings of the National Academy of Sciences of the United States of America, 118(5), e2019768118.
- Menon, U., et al. (2021). Ovarian cancer population screening and mortality after long-term follow-up in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. The Lancet, 397(10290), 2182–2193.
Topics: ctDNA, Methylated DNA